Lemtrada
BILAGA I
PRODUKTRESUMÉ
Detta läkemedel är föremål för utökad övervakning. Detta kommer att göra det möjligt att snabbt identifiera ny säkerhetsinformation. Hälso- och sjukvårdspersonal uppmanas att rapportera varje misstänkt biverkning. Se avsnitt 4.8 om hur man rapporterar biverkningar.
1. LÄKEMEDLETS NAMN
LEMTRADA 12 mg koncentrat till infusionsvätska, lösning.
2. KVALITATIV OCH KVANTITATIV SAMMANSÄTTNING
Varje injektionsflaska innehåller 12 mg alemtuzumab i 1,2 ml (10 mg/ml).
Alemtuzumab är en monoklonal antikropp som med hjälp av rekombinant DNA-teknik produceras i en suspensionskultur bestående av mammalieceller (äggstock från kinesisk hamster) och näringsmedel.
För fullständig förteckning över hjälpämnen, se avsnitt 6.1.
3. LÄKEMEDELSFORM
Koncentrat till infusionsvätska, lösning (sterilt koncentrat).
Ett klart, färglöst till svagt gulfärgat koncentrat med pH 7,0 - 7,4.
4. KLINISKA UPPGIFTER
4.1 Terapeutiska indikationer
LEMTRADA är indicerat för vuxna patienter med skovvis förlöpande multipel skleros (RRMS) med aktiv sjukdom som definieras av kliniska fynd eller bildfynd (se avsnitt 4.4 och 5.1).
4.2 Dosering och administreringssätt
Behandling med LEMTRADA bör initieras och övervakas av en neurolog med erfarenhet av behandling av MS-patienter. Specialister och utrustning som krävs för snabb diagnos och hantering av de vanligaste biverkningarna, särskilt autoimmuna tillstånd och infektioner, ska finnas tillgänglig.
Resurser för hantering av överkänslighets- och/eller anafylaktiska reaktioner ska finnas tillgänglig.
Patienter som behandlas med LEMTRADA måste förses med patientvarningskort och patientguide och informeras om riskerna med LEMTRADA (se även bipacksedeln).
Dosering
Den rekommenderade dosen av LEMTRADA är 12 mg/dag, administrerat genom intravenös infusion under två behandlingsomgångar.
• Initial behandlingsomgång: 12 mg/dag under fem på varandra följande dagar (total dos 60 mg)
• Andra behandlingsomgången: 12 mg/dag under tre på varandra följande dagar (total dos 36 mg), administrerat 12 månader efter den initiala behandlingsomgången.
Missade doser ska inte ges samma dag som en schemalagd dos.
Uppföljning av patienter
Behandlingen rekommenderas att ges i två behandlingsomgångar (se dosering) med säkerhetsuppföljning av patienterna från insättandet av behandlingen till och med 48 månader efter den sista infusionen (se avsnitt 4.4).
Premedicinering
Patienterna ska premedicineras med kortikosteroider omedelbart före administreringen av LEMTRADA under de första tre dagarna i varje behandlingsomgång. I de kliniska prövningarna premedicinerades patienterna med 1 000 mg metylprednisolon under de första tre dagarna av varje behandlingsomgång med LEMTRADA.
Utöver detta kan även premedicinering med antihistaminer och/eller antipyretika övervägas.
Oral profylax mot herpesinfektion ska administreras till alla patienter från och med den första dagen av varje behandlingsomgång och fortsätta i minst 1 månad efter behandlingen med LEMTRADA (se även "Infektioner" i avsnitt 4.4). I de kliniska prövningarna fick patienterna 200 mg aciklovir två gånger dagligen eller motsvarande.
Äldre
De kliniska studierna omfattade inga patienter över 55 års ålder. Det har därför inte fastställts om de svarar annorlunda på behandling jämfört med yngre patienter.
Nedsatt njur- eller leverfunktion
LEMTRADA har inte studerats på patienter med nedsatt njur- eller leverfunktion.
Pediatrisk population
Säkerhet och effekt för LEMTRADA för barn med MS i åldern 0 till 18 år har ännu inte fastställts. Det finns ingen relevant användning av alemtuzumab för behandling av multipel skleros hos barn i åldrarna nyfödda upp till 10 år. Inga data finns tillgängliga.
Administreringssätt
LEMTRADA måste spädas före infusion. Den utspädda lösningen ska administreras genom intravenös infusion under cirka 4 timmar.
Anvisningar om spädning av läkemedlet före administrering, se avsnitt 6.6.
4.3 Kontraindikationer
Överkänslighet mot den aktiva substansen eller mot något hjälpämne som anges i avsnitt 6.1.
Infektion med humant immunbristvirus (hiv).
4.4 Varningar och försiktighet
LEMTRADA rekommenderas inte till patienter med inaktiv sjukdom eller till de som är stabila på nuvarande behandling.
Patienter som behandlas med LEMTRADA måste få bipacksedeln, patientvarningskortet och patientguiden. Före behandlingen måste patienterna få information om risker och nytta samt det nödvändiga behovet av att genomföra 48 månaders uppföljning efter den sista infusionen av LEMTRADA.
Autoimmunitet
Behandling kan leda till att autoantikroppar bildas och ökar risken för autoimmunmedierade tillstånd, t.ex. immunologisk trombocytopeni (ITP), tyreoidearubbningar eller, i sällsynta fall, nefropatier (t.ex.
antiglomerulär basalmembranssjukdom). Försiktighet ska iakttas hos patienter med tidigare autoimmuna tillstånd andra än MS, även om tillgängliga data inte talar för någon försämring av tidigare autoimmuna tillstånd efter behandling med alemtuzumab.
Immunologisk trombocytopeni (ITP)
Allvarliga fall av ITP har observerats hos cirka 1 % av de MS-patienter som behandlats i kontrollerade kliniska prövningar. I en av dessa kontrollerade kliniska prövningar med patienter med MS, utvecklade en patient ITP som upptäcktes försent. Patienten avled av intracerebrala blödningar. Detta föranledde att krav på månatlig blodmonitorering infördes. ITP-debuten har i allmänhet skett mellan 14 och 36 månader efter den första exponeringen. Symtom på ITP kan omfatta (men är inte begränsade till) att patienten lätt får blåmärken, petekier, spontan mukokutan blödning (t.ex. epistaxis, hemoptys), kraftigare än normalt eller oregelbunden mensblödning. Hemoptys kan också vara indikativt för anti-GBM-sjukdom (se nedan) och en lämplig differentialdiagnos måste ställas. Påminn patienten om att vara fortsatt uppmärksam på eventuella symtom och vid minsta tveksamhet omedelbart uppsöka vård.
Fullständig blodstatus med differentialräkning ska mätas innan behandlingen påbörjas och därefter varje månad till och med 48 månader efter den sista infusionen. Efter denna tidsperiod ska provtagning utföras baserat på kliniska fynd som kan tyda på ITP. Om ITP misstänks ska fullständig blodstatus omedelbart mätas.
Om ITP-utveckling bekräftas ska lämplig medicinsk behandling omedelbart påbörjas, inklusive omedelbar remiss till specialist. Data från kliniska prövningar vid MS har visat att följsamhet till kraven på blodmonitorering och utbildning kring tecken och symtom på ITP har lett till tidig upptäckt och behandling av ITP, där de flesta fallen svarat på den medicinska förstahandsbehandlingen.
Den potentiella risken med fortsatt behandling med LEMTRADA efter förekomst av ITP är okänd.
Nefropatier
Nefropatier, inklusive anti-glomerulär basalmembranssjukdom (anti-GBM), har observerats hos 0,3 % av patienterna i kliniska prövningar vid MS och vanligen uppträtt inom 39 månader efter den sista administreringen av LEMTRADA. I de kliniska prövningarna inträffade två fall av anti-GBM-sjukdom.
Båda fallen var allvarliga, identifierades tidigt genom klinisk och laborativ monitorering och behandling utföll positivt.
Kliniska manifestationer av nefropati kan omfatta förhöjt serumkreatinin, hematuri och/eller proteinuri. Även om det inte observerats i kliniska prövningar, kan alveolär blödning manifesterad som hemoptys förekomma vid anti-GBM-sjukdom. Hemoptys kan också vara indikativt för ITP (se ovan) och en lämplig differentialdiagnos måste ställas. Patienten ska påminnas om att vara fortsatt uppmärksam på eventuella symtom och vid minsta tveksamhet omedelbart uppsöka vård. Anti-GBM-sjukdom kan leda till njursvikt som kräver dialys och/eller transplantation om den inte behandlas snabbt. Obehandlad kan den vara livshotande.
Serumkreatininnivåer ska mätas innan behandlingen påbörjas och, därefter varje månad till och med 48 månader efter den sista infusionen. Urinanalys med mikroskopi ska tas före insättning och därefter varje månad till och med 48 månader efter den sista infusionen. Vid en tydlig förändring av serumkreatinin jämfört med utgångsvärdet, oförklarlig hematuri och/eller proteinuri ska detta föranleda ytterligare utvärdering för nefropatier, inklusive omedelbar remiss till specialist. Tidig upptäckt och behandling av nefropatier kan minska risken för dåliga behandlingsresultat. Efter denna tidsperiod ska provtagning utföras baserat på kliniska fynd som kan tyda på nefropatier.
Den potentiella risken med fortsatt behandling med LEMTRADA efter förekomst av nefropatier är okänd.
Tyreoidearubbningar
Autoimmuna tyreoidearubbningar har observerats hos uppskattningsvis 36 % av patienterna som i kliniska prövningar behandlades med 12 mg LEMTRADA mot MS och följdes under 48 månader efter den första exponeringen för LEMTRADA. Incidensen för tyreoidearubbningar var högre hos patienter med tyreoidearubbningar i anamnesen, både i behandlingsgruppen med LEMTRADA och i behandlingsgruppen med interferon beta 1a (IFNB-1a). Hos patienter med pågående tyreoidearubbning bör LEMTRADA administreras om den möjliga nyttan överväger de möjliga riskerna. De observerade autoimmuna tyreoidearubbningarna inbegrep hypertyreos eller hypotyreos. De flesta fallen var av lindrig till måttlig svårighetsgrad. Före godkännandet av LEMTRADA inträffade allvarliga tyreoidearubbningar hos <1 % av patienterna och endast Basedows sjukdom (även känd som Graves sjukdom), hypertyreos och hypotyreos förekom hos fler än en patient. De flesta tyreoidearubbningarna hanterades med konventionell medicinsk behandling, men vissa patienter behövde genomgå kirurgiska ingrepp. I kliniska prövningar tilläts patienter som utvecklat tyreoidearubbningar fortsätta behandling med LEMTRADA. Även om erfarenheten är begränsad upplevde dessa patienter i regel inte någon försämring av tyreoideasjukdomens svårighetsgrad. Fortsatt behandling med LEMTRADA ska övervägas på individuell basis med hänsyn till det kliniska tillståndet hos respektive patient.
Funktionstester av sköldkörteln, såsom nivåer av tyreoideastimulerande hormon (TSH), ska mätas innan behandlingen påbörjas och därefter var 3 månad till och med 48 månader efter den sista infusionen. Därefter ska provtagning utföras baserat på kliniska fynd som kan tyda på tyreoidearubbning.
Tyreoideasjukdom innebär särskilda risker för gravida kvinnor (se avsnitt 4.6).
I de kliniska prövningarna var patientens antityreoideaperoxidas (anti-TPO)-antikroppsstatus före behandling inte indikativ för utvecklingen av en tyreoidearelaterad biverkning. Hälften av patienterna som testade positivt och en fjärdedel av patienterna som testade negativt för anti-TPO-antikroppar vid start utvecklade en tyreoidearubbning. Majoriteten (ungefär 80%) av patienterna som uppvisade en tyreoidearubbning efter behandling var negativa för anti-TPO-antikroppar vid start. Därför kan patienter utveckla en tyreoidearelaterad biverkning oavsett status för anti-TPO-antikroppar före behandling och måste därför genomgå alla tester med jämna mellanrum enligt beskrivningen ovan.
Cytopenier
Misstänkta autoimmuna cytopenier som neutropeni, hemolytisk anemi och pancytopeni har i sällsynta fall rapporterats i kliniska prövningar vid MS. Resultaten för fullständigt blodstatus (se ovan under ITP) ska användas för att övervaka cytopenier. Om cytopeni konstateras ska lämplig medicinsk behandling omedelbart påbörjas, inklusive remiss till specialist.
Infusionsrelaterade reaktioner (IAR:er (infusion associated reactions))
I kontrollerade kliniska studier har infusionsrelaterade reaktioner (IAR:er) definierats som en biverkning som inträffar under, eller inom 24 timmar efter, infusion med LEMTRADA. De flesta av dessa reaktioner kan bero på cytokinfrisättning under infusionen. De flesta patienter som behandlats med LEMTRADA i kontrollerade kliniska prövningar vid MS, upplevde milda till måttliga IAR:er under och/eller upp till 24 timmar efter administrering av 12 mg LEMTRADA. Reaktionerna inkluderade ofta huvudvärk, hudutslag, feber, illamående, urtikaria, klåda, sömnlöshet, frossa, flush, trötthet, andnöd, smakförändringar, obehag i bröstet, generaliserat utslag, takykardi, bradykardi, dyspepsi, yrsel och smärta. Allvarliga reaktioner förekom hos 3 % av patienterna, inklusive fall av feber, urtikaria, förmaksflimmer, illamående, obehag i bröstet och hypotoni. Kliniska manifestationer av anafylaxi kan likna de kliniska manifestationerna för infusionsrelaterade reaktioner, men de är ofta allvarligare eller potentiellt livshotande. Rapporter om reaktioner som tillskrivs anafylaxi är sällsynta, till skillnad från infusionsrelaterade reaktioner.
Det rekommenderas att patienterna premedicineras för att lindra effekterna av infusionsreaktioner (se avsnitt 4.2). De flesta patienterna i de kontrollerade kliniska prövningarna fick antihistaminer och/eller antipyretika före minst en infusion med LEMTRADA. IAR:er kan inträffa hos patienterna trots premedicinering. Monitorering av infusionsreaktioner rekommenderas under infusion med LEMTRADA samt 2 timmar efter. Om en IAR uppstår, ge vid behov lämplig symtomatisk behandling. Om infusionen inte tolereras väl kan infusionstiden förlängas. Om allvarliga infusionsrelaterade reaktioner inträffar ska omedelbart utsättande av den intravenösa infusionen övervägas. I de kliniska prövningarna var anafylaxi eller allvarliga reaktioner som ledde till att behandlingen avslutades mycket sällsynta. Läkare ska känna till patientens hjärtanamnes eftersom infusionsrelaterade reaktioner kan omfatta hjärtsymtom så som takykardi.
Resurser för hantering av anafylaktiska eller allvarliga reaktioner måste finnas tillgängliga. Infektioner
I kontrollerade kliniska prövningar vid MS med upp till 2 års varaktighet förekom infektioner hos 71 % av patienterna som behandlades med LEMTRADA 12 mg jämfört med 53 % av patienterna behandlade med subkutant interferon beta-1a [IFNB 1a] (44 mikrogram 3 gånger i veckan), och var övervägande av mild till måttlig svårighetsgrad. Infektioner som förekom oftare hos LEMTRADA-behandlade patienter än patienter som behandlats med IFNB 1a inkluderade nasofaryngit, urinvägsinfektion, övre luftvägsinfektion, bihåleinflammation, munherpes, influensa och bronkit. Allvarliga infektioner förekom hos 2,7 % av patienterna som behandlades med LEMTRADA jämfört med 1 % av patienterna som behandlades med IFNB-1a i kontrollerade kliniska prövningar vid MS. Allvarliga infektioner i LEMTRADA-gruppen omfattade: blindtarmsinflammation, gastroenterit, pneumoni, herpes zoster och tandinfektion. Infektionerna hade i allmänhet ordinär varaktighet och försvann efter konventionell medicinsk behandling.
Allvarliga infektioner med varicella zoster-virus, inklusive primär varicella och varicella zoster-reaktivering, inträffade oftare hos patienter som behandlats med LEMTRADA 12 mg (0,3 %) i kliniska prövningar, jämfört med IFNB-1a (0 %). Infektion med cervikalt humant papillomvirus (HPV), inklusive cervixdysplasi, har också rapporterats hos patienter som behandlats med LEMTRADA 12 mg (2 %). Det rekommenderas att HPV-screening genomförs årligen för kvinnliga patienter.
Tuberkulos har rapporterats hos patienter som behandlats med LEMTRADA och IFNB-1a i kontrollerade kliniska prövningar. Aktiv och latent tuberkulos har rapporterats hos 0,3 % av patienterna som behandlats med LEMTRADA, oftast i endemiska områden. Innan behandling sätts in måste alla patienter utvärderas för både aktiv och inaktiv (”latent”) tuberkulosinfektion, enligt lokala riktlinjer.
Listerios/listeria-meningit har rapporterats hos patienter som behandlats med LEMTRADA, vanligtvis inom en månad efter infusion. För att minska denna risk bör patienter som får LEMTRADA undvika intag av rått eller dåligt tillagat kött, mögel- och kittost och opastöriserade mejeriprodukter i minst en månad efter LEMTRADA-behandling.
Ytliga svampinfektioner, särskilt oral och vaginal candida-infektion, inträffade oftare hos LEMTRADA-behandlade patienter (12 %) än hos patienter som behandlats med IFNB-1a (3 %) i kontrollerade kliniska prövningar vid MS.
Läkare bör överväga att senarelägga initieringen av administrering av LEMTRADA till patienter med aktiv infektion, till dess infektionen är helt under kontroll.
Profylax med oralt antiherpesmedel bör sättas in till alla patienter från och med den första dagen av behandling med LEMTRADA och fortsätta i minst 1 månad efter varje behandlingsomgång. I kliniska prövningar fick patienterna 200 mg aciklovir två gånger dagligen eller motsvarande.
LEMTRADA har inte administreras för behandling av MS samtidigt som eller efter antineoplastiska eller immunsuppressiva behandlingar. Som med andra immunmodulerande behandlingar ska potentiella kombinationseffekter på patientens immunsystem tas med i beräkningen när administrering av LEMTRADA övervägs. Samtidig användning av LEMTRADA med någon av dessa behandlingar kan öka risken för immunsuppression.
Det finns inga uppgifter om samband mellan LEMTRADA och reaktivering av hepatit B-virus (HBV) eller hepatit C-virus (HCV) eftersom patienter med tecken på aktiva eller kroniska infektioner uteslöts ur de kliniska prövningarna. Screening av patienter med hög risk för HBV- och/eller HCV-infektion bör övervägas före behandling med LEMTRADA, och försiktighet bör iakttas vid förskrivning av LEMTRADA till patienter som bär på HBV och/eller HCV eftersom dessa patienter kan vara i riskzonen för bestående leverskada relaterad till en potentiell virusreaktivering till följd av den tidigare virusinfektionen.
Malignitet
Som med andra immunmodulerande terapier ska försiktighet iakttas vid insättning av LEMTRADA hos patienter med tidigare och/eller pågående malignitet. Det är i dagsläget inte känt om alemtuzumab medför en högre risk för att utveckla maligniteter i tyreoidea, eftersom tyreoidautoimmunitet i sig kan utgöra en riskfaktor för maligniteter i tyreoidea.
Preventivmedel
Överföring via placenta och potentiell farmakologisk aktivitet av LEMTRADA observerades hos möss under dräktighet och efter födseln. Kvinnor i fertil ålder bör använda effektiva preventivmedel under behandlingen samt under 4 månader efter en behandlingsomgång med LEMTRADA (se avsnitt 4.6).
Vacciner
Det rekommenderas att patienterna har slutfört lokala vaccineringskrav minst 6 veckor före behandling med LEMTRADA. Förmågan att generera ett immunsvar mot något vaccin efter behandling med LEMTRADA har inte studerats.
Säkerheten vid immunisering med levande virusvacciner efter en behandlingsomgång med LEMTRADA har inte formellt studerats vid kontrollerade kliniska prövningar vid MS, och ska inte administreras till MS-patienter som nyligen har fått en behandlingsomgång med LEMTRADA.
Test av/vaccinering mot varicella zoster-virusantikroppar
Som för alla immunmodulerande läkemedel bör patienter utan anamnes på vattkoppor eller utan vaccination mot varicella zoster-virus (VZV) testas för antikroppar mot VZV innan behandling med LEMTRADA påbörjas. VZV-vaccinering av antikroppsnegativa patienter bör övervägas innan behandling med LEMTRADA inleds. För full effekt av VZV-vaccination ska behandling med LEMTRADA skjutas upp till 6 veckor efter vaccinationen.
Rekommenderade laboratorietester för patientmonitorering
Laboratorietester skall utföras med jämna mellanrum under 48 månader efter den sista behandlingen med LEMTRADA för att övervaka tidiga tecken på autoimmun sjukdom:
• Fullständigt blodstatus med differentialräkning (före behandlingsstart och därefter varje månad)
• Serumkreatininnivåer (före behandlingsstart och därefter varje månad)
• Urinanalys med mikroskopi (före behandlingsstart och därefter varje månad)
• Test av tyreoideafunktionen, såsom nivån av tyreoideastimulerande hormon (TSH) (före behandlingsstart och därefter var tredje månad)
Efter denna tidsperiod föranleder alla kliniska fynd som tyder på nefropati eller tyreoidearubbning ytterligare provtagning.
Information från utanför av företaget sponsrade studier vad gäller användning av alemtuzumab före godkännandet för försäljning av LEMTRADA
Följande biverkningar har identifierats före registreringen av LEMTRADA under användning av alemtuzumab för behandling av kronisk lymfatisk B-cellsleukemi (B-KLL), samt för behandling av andra sjukdomar, i allmänhet vid högre och tätare dosering (t.ex. 30 mg) än vad som rekommenderas för behandling av MS. Eftersom dessa biverkningar har rapporterats frivilligt från en population av obestämd storlek är det inte alltid möjligt att med säkerhet beräkna frekvensen eller fastställa ett orsakssamband med exponering för alemtuzumab.
Autoimmun sjukdom
Autoimmuna händelser som rapporterats hos alemtuzumab-behandlade patienter omfattar neutropeni, hemolytisk anemi (inklusive ett dödligt fall), förvärvad hemofili, anti-GBM-sjukdom och tyreoideasjukdom. Allvarliga och ibland dödliga autoimmuna tillstånd inklusive autoimmun hemolytisk anemi, autoimmun trombocytopeni, aplastisk anemi, Guillain-Barrés syndrom och kronisk inflammatorisk demyeliniserande polyradikuloneuropati har rapporterats hos alemtuzumab-behandlade icke-MS-patienter. Ett positivt Coombs-test har rapporterats hos en alemtuzumab-behandlad cancerpatient. Ett dödsfall i samband med transfusionsrelaterad transplantat-mot-värd-reaktion har rapporterats hos en alemtuzumab-behandlad cancerpatient.
Infusionsrelaterade reaktioner
Allvarliga och ibland dödliga infusionsrelaterade reaktioner inklusive bronkospasm, hypoxi, synkope, lunginfiltrat, akut respiratorisk insufficiens, andningsstillestånd, hjärtinfarkt, arytmier, akut hjärtinsufficiens och hjärtstillestånd har observerats hos icke-MS-patienter som behandlats med alemtuzumab med högre och mer frekventa doser än vad som används vid MS. Svår anafylaxi och andra överkänslighetsreaktioner, inklusive anafylaktisk chock och angioödem har också rapporterats.
Infektioner och infestationer
Allvarliga och ibland dödliga virus, bakterier, protozoer, och svampinfektioner, inklusive sådana som beror på reaktivering av latenta infektioner, har rapporterats hos icke-MS-patienter som behandlats med alemtuzumab med högre och tätare doser än vad som används vid MS. Progressiv multifokal leukoencefalopati (PML) har rapporterats hos patienter med B-KLL med eller utan behandling med alemtuzumab. Frekvensen av PML hos B-KLL-patienter som behandlats med alemtuzumab är inte större än hos normalpopulationen.
Blodet och lymfsystemet
Allvarliga blödningsreaktioner har rapporterats hos icke-MS-patienter.
Hjärtat
Hjärtsvikt, kardiomyopati och minskad ejektionsfraktion har rapporterats hos alemtuzumab-behandlade icke-MS-patienter som tidigare behandlats med potentiellt kardiotoxiska medel.
Epstein-Barr virusassocierade lymfoproliferativa sjukdomar Epstein-Barr virusassocierade lymfoproliferativa sjukdomar har observerats.
4.5 Interaktioner med andra läkemedel och övriga interaktioner
Inga formella interaktionsstudier har utförts för LEMTRADA med den rekommenderade dosen för patienter med MS. I en kontrollerad klinisk studie på MS-patienter som nyligen behandlats med beta-interferon och glatirameracetat krävdes att behandlingen avslutades 28 dagar innan behandling med LEMTRADA påbörjades.
4.6 Fertilitet, graviditet och amning
Kvinnor i fertil ålder
Serumkoncentrationerna av alemtuzumab var låga eller icke detekterbara inom cirka 30 dagar efter varje behandlingsomgång. Kvinnor i fertil ålder bör därför använda effektiva preventivmedel under en behandlingsomgång med LEMTRADA samt under 4 månader efter denna behandlingsomgång.
Graviditet
Det finns begränsat med data från användning av LEMTRADA hos gravida kvinnor. LEMTRADA ska administreras under graviditet endast då de potentiella fördelarna överväger den eventuella risken för fostret.
Det är känt att humant IgG passerar placentabarriären. Alemtuzumab skulle därför kunna passera placentabarriären och eventuellt utgöra en risk för fostret. Djurstudier har visat reproduktionstoxikologiska effekter (se avsnitt 5.3). Det är inte känt om alemtuzumab kan orsaka fosterskador när det ges till gravida kvinnor eller om det kan påverka reproduktionsförmågan.
Tyreoidearubbningar (se avsnitt 4.4 Tyreoidearubbningar) innebär särskilda risker för gravida kvinnor. Vid obehandlad hypotyreos under graviditet föreligger en ökad risk för missfall och skador på fostret, såsom mental retardation och dvärgväxt. Hos mödrar med Graves sjukdom kan tyreoideastimulerande hormonreceptorantikroppar hos modern överföras till ett växande foster och orsaka övergående neonatal Graves sjukdom.
Amning
Alemtuzumab har detekterats i mjölk och hos avkomma från ammande honmöss.
Det är okänt om alemtuzumab utsöndras i bröstmjölk. En risk för det ammande barnet kan inte uteslutas. Amning bör därför avbrytas under varje behandlingsomgång med LEMTRADA samt under 4 månader efter den sista infusionen i varje behandlingsomgång. Dock kan fördelarna med att få immunitet genom bröstmjölk uppväga riskerna för potentiell exponering för alemtuzumab för det ammande barnet.
Fertilitet
Det finns inte tillräckliga kliniska säkerhetsdata om effekterna av LEMTRADA på fertilitet. I en delstudie på 13 manliga patienter behandlade med alemtuzumab (som behandlats med antingen 12 mg eller 24 mg) sågs inga tecken på aspermi, azoospermi, bestående minskat antal spermier, störningar av motilitet eller en ökning av morfologiska abnormiteter hos spermierna.
Det är känt att CD52 förekommer i reproduktiv vävnad hos människor och gnagare. Djurdata har visat effekter på fertiliteten hos humaniserade möss (se avsnitt 5.3), men baserat på tillgängliga data är en potentiell påverkan på humanfertilitet under exponeringen okänd.
4.7 Effekter på förmågan att framföra fordon och använda maskiner
Inga studier har utförts avseende effekterna av LEMTRADA på förmågan att framföra fordon eller använda maskiner.
De flesta patienter får IAR:er som uppkommer under, eller inom 24 timmar efter, behandling med LEMTRADA. Vissa IAR:er (t.ex. yrsel) kan tillfälligt påverka patientens förmåga att framföra fordon eller använda maskiner och försiktighet ska iakttas tills dessa reaktioner har gått över.
4.8 Biverkningar
Sammanfattning av säkerhetsprofilen
Totalt 1 188 patienter med skovvis förlöpande MS (RRMS) som behandlats med LEMTRADA (12 mg eller 24 mg) utgjorde säkerhetspopulationen vid en poolad analys av kontrollerade kliniska studier, vilket resulterade i 2 363 patientår av säkerhetsuppföljning med en medianuppföljningstid på 24 månader.
De viktigaste biverkningarna var autoimmunitet (ITP, tyreoidearubbningar, nefropatier, cytopenier), IAR:er och infektioner. Dessa beskrivs i avsnitt 4.4.
De vanligaste biverkningarna av LEMTRADA (hos >20% av patienterna) var hudutslag, huvudvärk, feber och luftvägsinfektioner.
Lista över biverkningar
Nedanstående tabell är baserad på poolade säkerhetsdata upp till 24 månader från RRMS-patienter som behandlats med LEMTRADA 12 mg/dag under fem på varandra följande dagar vid studiens början och under tre på varandra följande dagar i studiemånad 12. Biverkningar som förekom hos > 0,5% av patienterna anges efter Medical Dictionary for Regulatory Activities (MedDRA), systemorgansystem (SOC) och föredragen term (PT (preferred term)). Frekvenserna definieras enligt följande: Mycket vanliga (> 1/10), vanliga (> 1/100 till < 1/10), mindre vanliga (> 1/1 000 till < 1/100). Biverkningarna presenteras inom varje frekvensområde efter fallande allvarlighetsgrad.
Tabell 1: Biverkningar i studie 1, 2 och 3 som observerats hos >0,5% av patienter som behandlats med LEMTRADA 12 mg
|
Organklass |
Mycket vanliga |
Vanliga |
Mindre vanliga |
|
Infektioner och infestationer |
Övre luftvägsinfektion, urinvägsinfektion |
Nedre luftvägsinfektioner, herpes zoster, gastroenterit, oral herpes, oral candida, vulvovaginal candida, influensa, öroninfektion |
Tandinfektion, genital herpes, onykomykos |
|
Blodet och lymfsystemet |
Lymfopeni, leukopeni |
Lymfadenopati |
Immun trombocytopen purpura, trombocytopeni, minskat hemoglobin, minskad hematokrit |
|
Immunsystemet |
Cytokinfrisättningssyndro m | ||
|
Endokrina systemet |
Basedows sjukdom, hypertyreos, autoimmun tyroidit, hypotyreos, struma, anti-tyreoid-antikropps-positiv | ||
|
Psykiska störningar |
Sömnlöshet*, ångest |
Depression | |
|
Centrala och perifera nervsystemet |
Huvudvärk* |
MS-skov, yrsel*, hypestesi, parestesi, tremor, dysgeusi* |
Sensorisk störning, hyperestesi |
|
Ögon |
Dimsyn |
Konjunktivit | |
|
Öron och balansorgan |
Svindel | ||
|
Hjärtat |
Tachykardi*, bradykardi*, palpitationer | ||
|
Blodkärl |
Flush* |
Hypotoni*, hypertoni | |
|
Andningsvägar, bröstkorg och mediastinum |
Dyspné*, hosta, näsblod, orofaryngeal smärta |
Trånghetskänsla i svalget, hicka, halsirritation | |
|
Magtarmkanalen |
Illamående* |
Buksmärtor, kräkningar, diarré, dyspepsi*, stomatit |
Förstoppning, gastroesofageal reflux, gingival blödning, dysfagi |
|
Lever och gallvägar |
Förhöjt aspartataminotransferas (ASAT) | ||
|
Hud och subkutan vävnad |
Urtikaria*, hudutslag*, klåda* |
Generaliserade utslag *, erytem,ekkymos, alopeci, hyperhidros, akne |
Blåsor, nattliga svettningar |
|
Muskuloskeletala systemet och bindväv |
Myalgi, muskelsvaghet, ledvärk, ryggsmärtor, smärta i extremiteter, muskelspasmer, nacksmärta | ||
|
Njurar och urinvägar |
Proteinuri, hematuri | ||
|
Reproduktionsorgan och bröstkörtel |
Menorragi, oregelbunden menstruation |
Cervikal dysplasi, amenorré |
|
Allmänna symtom och/eller symtom vid administreringsstället |
Feber*, trötthet* |
Obehag i bröstet*, frossa*, smärta*, perifert ödem, asteni, influensaliknande sjukdom, sjukdomskänsla, smärta vid infusionsstället | |
|
Undersökningar |
Viktnedgång | ||
|
Skador, förgiftningar och behandlingskomplikat ioner |
Blåmärke |
Beskrivning av utvalda biverkningar
Termer som är markerade med asterisk (*) i tabell 1 innefattar biverkningar som rapporterats som infusionsrelaterade reaktioner. IAR: er omfattar även förmaksflimmer och anafylaxi, som inträffat under 0,5%-nivån för relaterade reaktioner (se avsnitt 4.4).
Rapportering av misstänkta biverkningar
Det är viktigt att rapportera misstänkta biverkningar efter att läkemedlet godkänts. Det gör det möjligt att kontinuerligt övervaka läkemedlets nytta-riskförhållande. Hälso- och sjukvårdspersonal uppmanas att rapportera varje misstänkt biverkning via det nationella rapporteringssystemet listat i bilaga V.
4.9 Överdosering
I kontrollerade kliniska prövningar fick två MS-patienter av misstag upp till 60 mg LEMTRADA (dvs. total dos under den inledande behandlingsomgången) i en enda infusion och upplevde allvarliga reaktioner (huvudvärk, hudutslag och antingen hypotoni eller sinustakykardi). Större doser av LEMTRADA än de som testats i kliniska studier kan öka intensiteten och/eller varaktigheten av infusionsrelaterade biverkningar eller dess effekter på immunförsvaret.
Det finns ingen känd antidot mot överdosering av alemtuzumab. Behandlingen består av utsättande av läkemedlet och symtomatisk behandling.
5. FARMAKOLOGISKA EGENSKAPER
5.1 Farmakodynamiska egenskaper
Farmakoterapeutisk grupp: Selektiva immunsuppressiva medel, ATC-kod: L04AA34.
Verkningsmekanism
Alemtuzumab är en rekombinant, DNA-deriverad humaniserad monoklonal antikropp som är riktad mot cellytan på 21-28 kD glykoprotein CD52. Alemtuzumab är en IgG1-kappa-antikropp med humant variabelt ramverk och konstanta regioner samt komplementära styrande regioner från en murin (råtta) monoklonal antikropp. Antikroppen har en molekylvikt på cirka 150 kD.
Alemtuzumab binder till CD52, ett antigen som förekommer i höga nivåer på cellytan på T- (CD3+) och B-(CD19+) lymfocyter och i lägre nivåer på NK-celler, monocyter och makrofager. Få eller inga CD52 har upptäckts på neutrofiler, plasmaceller eller stamceller från benmärg. Alemtuzumab agerar genom antikroppsberoende cellulär cytolys och komplementförmedlad lysering efter att ha bundit till cellytan på T-och B-lymfocyter.
Den mekanism genom vilken LEMTRADA utövar dess terapeutiska effekter vid MS är inte helt klarlagd.
Forskningen tyder emellertid på immunmodulerande effekter genom utarmning och återbildning av lymfocyter, inklusive:
- Förändringar av antalet, proportionerna och egenskaperna hos vissa undergrupper av lymfocyter efter behandling
- Ökad förekomst av regulatoriska T-cellsundergrupper
- Ökad förekomst av minnes-T- och B-lymfocyter
- Övergående effekter på delar av den medfödda immuniteten (dvs neutrofiler, makrofager, NK-celler)
Minskningen av nivån av cirkulerande B-och T-celler till följd av LEMTRADA-behandling och den efterföljande återbildningen av immunceller kan minska risken för skov, vilket i slutänden kan fördröja sjukdomsförloppet.
Farmakodynamisk effekt
LEMTRADA utarmar cirkulerande T- och B-lymfocyter efter varje behandlingsomgång, med de lägsta observerade värdena en månad efter en behandlingsomgång (den tidigaste tidpunkten efter behandlingen i fas 3-studierna). Lymfocyter återbildas över tiden och B-cellsåterhämtningen är vanligtvis avslutad inom 6 månader. Antalet CD3+- och CD4+-lymfocyter stiger långsammare mot normalvärdena, och har i allmänhet inte återgått till ursprungsnivåerna 12 månader efter behandlingen. Cirka 40% av patienterna hade totala lymfocytantal som nådde den nedre gränsen för normalvärde (LLN) 6 månader efter varje behandlingsomgång och cirka 80% av patienterna hade totala lymfocytantal som nådde LLN 12 månader efter varje omgång.
Neutrofiler, monocyter, eosinofiler, basofiler och NK-celler påverkas endast övergående av LEMTRADA. Klinisk effekt och säkerhet
Säkerheten och effekten av LEMTRADA har utvärderats i tre randomiserade, bedömarblindade (rater blinded) kliniska prövningar med jämförelse mot aktivt behandlade patienter med RRMS.
För studierna 1 och 2 anges studiens uppläggning/demografi och resultat i tabell 2 respektive tabell 3.
|
Tabell 2: Studieuppläggning och egenskaper vid start för studie 1 och 2 | ||
|
Studie 1 |
Studie 2 | |
|
Studiens namn |
CAMMS323 (CARE-MS I) |
CAMMS32400507 (CARE-MS II) |
|
Studiens uppläggning | ||
|
Sjukdomshistoria |
Patienter med aktiv MS , definierat som minst 2 skov under de föregående 2 åren. | |
|
Uppföljning |
2 år | |
|
Studiepopulation |
Behandlingsnaiva patienter |
Patienter med inadekvat respons på tidigare behandling* |
|
Egenskaper vid start av studie | ||
|
Medelålder (år) |
33 |
35 |
|
Genomsnittlig /median varaktighet på sjukdomen |
2,0/1,6 år |
4,5/3,8 år |
|
Genomsnittlig varaktighet av tidigare MS-behandling (>1 läkemedel använt) |
Ingen |
36 månader |
|
% som fått >2 tidigare MS-behandlingar |
Ej relevant |
28% |
|
Genomsnittlig EDSS-poäng vid start av studien |
2,0 |
2,7 |
* Definierat som patienter som har upplevt minst ett skov under behandling med beta-interferon eller glatirameracetat efter att ha fått behandling med läkemedel under minst sex månader.
|
Tabell 3: Viktiga kliniska och MRI-endpoints från studie 1 och 2 | ||||
|
Studie 1 |
Studie 2 | |||
|
Studiens namn |
CAMMS323 (CARE-MS I) |
CAMMS32400507 (CARE-MS II) | ||
|
Kliniska endpoints |
LEMTRADA 12 mg (N=376) |
SC IFNB-1a (N=187) |
LEMTRADA 12 mg (N=426) |
SC IFNB-1a (N=202) |
|
Skovfrekvens1 ARR (Annualised relapse rate) (95% CI) |
0,18 (0,13, 0,23) |
0,39 (0,29, 0,53) |
0,26 (0,21, 0,33) |
0,52 (0,41, 0,66) |
|
Frekvenskvot (rate ratio) (95 % CI) Riskminskning |
0,45 (0,32, 0,63) 54,9 (p<0,0001) |
0,51 (0,39, 0,65) 49,4 (p<0,0001) | ||
|
Funktionsnedsättning2 (SAD [Sustained accumulation of disability] >6 månader1) Patienter med SAD vid 6 månader (95% CI) |
8,0% (5,7, 11,2) |
11,1% (7,3, 16,7) |
12,7% (9,9, 16,3) |
21,1% (15,9, 27,7) |
|
Relativ risk (hazard ratio) (95% CI) |
0,70 (0,40, 1,23) (p=0,22) |
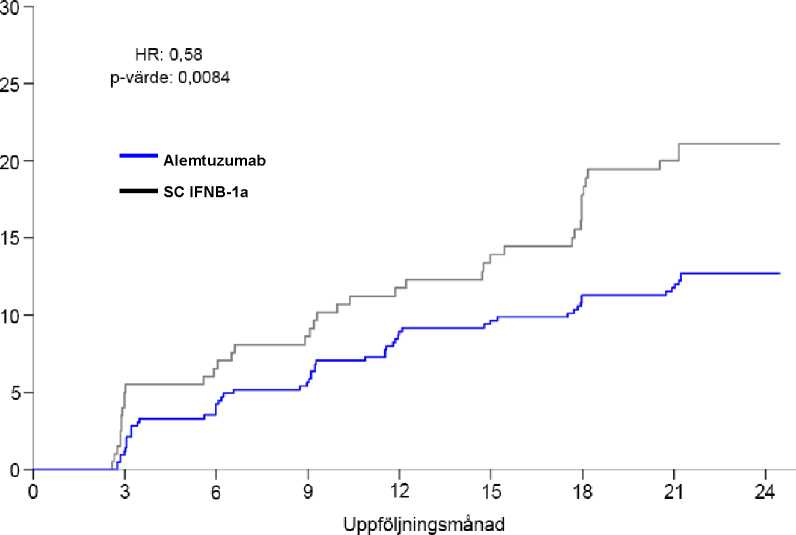
0,58 (0,38, 0,87) (p=0,0084) | ||
|
Patienter som var skovfria år 2 (95% CI) |
77,6% (72,9, 81,6) (p<0,0001) |
58,7% (51,1, 65,5) |
65,4% (60,6, 69,7) (p<0,0001) |
46,7% (39,5, 53,5) |
|
Förändring från studiestart av EDSS år 2 Uppskattning (95% CI) |
-0,14 (-0,25, -0,02) (p=0,42) |
-0,14 (-0,29, 0,01) |
-0,17 (-0,29, -0,05) (p<0,0001) |
0,24 (0,07, 0,41) |
|
MRI-endpoints (0-2 år) | ||||
|
Medianförändring (%) av MRI-T2-lesionsvolym |
-9,3 (-19,6, -0,2) (p=0,31) |
-6,5 (-20,7, 2,5) |
-1,3 (p=0,14) |
-1,2 |
|
Patienter med nya eller förstorade T2-lesioner under år 2 |
48,5% (p=0,035) |
57,6% |
46,2% (p<0,0001) |
67,9% |
|
Patienter med gadoliniumförstärkta lesioner under år 2 |
15,4% (p=0,001) |
27,0% |
18,5% (p<0,0001) |
34,2% |
|
Patienter med nya hypointensiva Tl-lesioner under år 2 |
24,0% (p=0,055) |
31,4% |
19,9% (p<0,0001) |
38,0% |
|
Medianförändring (%) av hjärnans parenkymala fraktion |
-0,867 (p<0,0001) |
-1,488 |
-0,615 (p=0,012) |
-0,810 |
|
1 Co-primära endpoints: ARR och SAD. Om minst en av två co-primära endpoints uppfylldes ansågs studien uppfylla sitt mål. 2 Tid till SAD (sustained accumulation of disability)definierades som en ökning med minst 1 poäng på Expanded Disability Status Scale (EDSS) om EDSS-poängen vid start var >1,0 (1,5 poängs ökning för patienter med en EDSS vid start = 0) och som kvarstod i 6 månader. | ||||
Figur 1: Tid till 6-månaders SAD i studie 2
Q
<
CO
~o
CD
E
aj
c
.CD
M
Q.
"cd
T3
C
ra
"c
CD
O
O
it
Skovens svårighetsgrad
I linje med effekten på skovfrekvensen visade stödjande analyser från Studie 1 (CAMMS323) att LEMTRADA 12 mg/dag ledde till att signifikant färre LEMTRADA-behandlade patienter upplevde allvarliga skov (61% minskning, p = 0,0056) och signifikant färre skov som ledde till steroidbehandling (58% minskning, p <0,0001) jämfört med IFNB-1a.
Stödjande analyser från Studie 2 (CAMMS32400507) visade att LEMTRADA 12 mg/dag ledde till att signifikant färre patienter behandlade med LEMTRADA upplevde svåra skov (48% minskning, p = 0,0121) och signifikant färre skov ledde till steroidbehandling (56 % minskning, p <0,0001) eller till sjukhusvistelse (55% minskning, p = 0,0045) jämfört med IFNB-1a.
Bestående minskning av funktionshinder (SRD (sustained reduction of disability))
Tid till SRD-debut definierades som en minskning med minst 1 poäng av EDSS från en EDSS-poäng vid studiestart >2 och som kvarstod i minst sex månader. SRD är ett mått på bestående förbättring av funktionsnedsättning. 29% av patienterna som behandlades med LEMTRADA nådde SRD i studie 2, medan endast 13% av de subkutant IFNB-1a-behandlade patienterna nådde denna endpoint. Skillnaden var statistiskt signifikant (p=0,0002).
Studie 3 (fas 2-studien CAMMS223) utvärderade säkerheten och effekten av LEMTRADA på patienter med RRMS under loppet av 5 år. Patienterna hade en EDSS från 0-3,0, minst två kliniska skov av MS under de föregående två åren och >1 gadoliniumförstärkt lesion vid studiens början. Patienterna hade inte tidigare fått behandling för MS. Patienterna behandlades med LEMTRADA 12 mg/dag (N = 108) eller 24 mg/dag (n = 108) vilket administrerades en gång per dag under fem dagar vid månad 0 och under tre dagar vid månad 12 eller subkutant IFNB-1a 44 pg (N = 107) administrerat tre gånger per vecka under tre år. Fyrtiosex patienter fick en tredje behandlingsomgång med LEMTRADA med 12 mg/dag eller 24 mg/dag under tre dagar vid månad 24.
Vid år tre minskade LEMTRADA risken för 6-månaders-SAD med 76% (riskkvot 0,24 [95% CI: 0,110, 0,545], p<0,0006) och reducerade ARR med 67% (frekvenskvot 0,33 [95% CI: 0,196, 0,552], p<0,0001) jämfört med subkutant IFNB-1a. Alemtuzumab 12 mg/dag ledde till signifikant lägre EDSS-poäng (förbättrats från studiestart) under två års uppföljning jämfört med IFNB-1a (p <0,0001).
Vid 5 år minskade LEMTRADA risken för SAD med 69% (riskkvot 0,31 [95% CI: 0,161, 0,598], p=0,0005) och reducerade ARR med 66 % (frekvenskvot 0,34 [95% CI: 0,202, 0,569], p<0,0001) jämfört med subkutant IFNB-1a.
I en öppen uppföljningsstudie av de kliniska prövningarna av LEMTRADA fick vissa patienter ytterligare behandling "vid behov" med LEMTRADA efter dokumenterade tecken på återfall av MS-sjukdomsaktivitet. Den/de extra behandlingsomgången/arna med LEMTRADA administrerades med 12 mg/dag under tre på varandra följande dagar (total dos 36 mg), minst 12 månader efter den föregående behandlingsomgången. Fördelarna och riskerna med >2 behandlingsomgångar har inte helt fastställts, men resultaten tyder på att säkerhetsprofilen inte verkar förändras med ytterligare behandlingsomgångar. Om ytterligare behandlingsomgångar ska ges måste de administreras minst 12 månader efter föregående omgång.
Immunogenicitet
I likhet med alla terapeutiska proteiner finns det risk för immunogenicitet. Data återspeglar andelen patienter vars testresultat ansågs positiva för antikroppar mot alemtuzumab med användning av en enzymkopplad immunabsorberande analys (ELISA) och bekräftades genom en kompetitiv bindningsanalys. Positiva prover utvärderades ytterligare med avseende på tecken på in vitro-hämning genom användning av en flödescytometrianalys. Patienter i kontrollerade kliniska prövningar vid MS fick serumprover tagna en, tre och 12 månader efter varje behandlingsomgång för att bestämma förekomsten av anti-alemtuzumab-antikroppar. Cirka 85% av patienterna som fick LEMTRADA testade positivt för anti-alemtuzumab-antikroppar under studien, och 92% av dessa patienter testade även positivt för antikroppar som hämmade LEMTRADA-bindning in vitro. De patienter som utvecklade anti-alemtuzumab-antikroppar gjorde detta vid 15 månader efter första exponeringen. Det fanns inget samband mellan förekomsten av anti-alemtuzumab-eller hämmande anti-alemtuzumab-antikroppar och en minskning av effekt, förändrad farmakodynamik eller förekomsten av biverkningar, inklusive infusionsrelaterade reaktioner.
Incidensen av antikroppar är starkt beroende av analysens känslighet och specificitet. Dessutom kan den observerade incidensen av antikroppar (inklusive hämmande antikroppar) i en analys påverkas av flera faktorer, inklusive analysmetod, provhantering, tidpunkten för provtagning, samtidig medicinering och underliggande sjukdom. Av dessa skäl kan jämförelse av incidensen av antikroppar mot LEMTRADA med incidensen av antikroppar mot andra produkter vara vilseledande.
Pediatrisk population.
Europeiska läkemedelsmyndigheten har beviljat undantag från kravet att skicka in studieresultat för alemtuzumab för behandling av multipel skleros hos barn i åldrarna nyfödda upp till 10 år (information om pediatrisk användning finns i avsnitt 4.2).
Europeiska läkemedelsmyndigheten har senarelagt kravet att skicka in studieresultat för LEMTRADA för en eller flera grupper av den pediatriska populationen för RRMS (information om pediatrisk användning finns i avsnitt 4.2).
5.2 Farmakokinetiska egenskaper
Farmakokinetiken för LEMTRADA utvärderades på sammanlagt 216 patienter med RRMS som fick intravenösa infusioner med antingen 12 mg/dag eller 24 mg/dag under fem på varandra följande dagar, följt av tre på varandra följande dagar 12 månader efter den första behandlingsomgången. Serumkoncentrationer ökade med varje dos under en behandlingsomgång, med de högsta observerade koncentrationerna efter den sista infusionen under en behandlingsomgång. Administration av 12 mg/dag resulterade i en genomsnittlig Cmax på 3014 ng/ml dag fem i den initiala behandlingsomgången och 2276 ng/ml dag tre i den andra behandlingsomgången. Alfa-halveringstiden uppskattades till fyra till fem dagar och var jämförbar mellan behandlingsomgångama och ledde till låga eller icke detekterbara serumkoncentrationer inom cirka 30 dagar efter varje omgång.
Alemtuzumab är ett protein för vilket den förväntade metaboliska vägen är nedbrytning till små peptider och enskilda aminosyror genom brett distribuerade proteolytiska enzymer. Inga klassiska biotransformationsstudier har utförts.
Utifrån tillgänglig data kan inga slutsatser dras om farmakokinetiken för LEMTRADA med avseende på effekten av etnicitet och kön. Farmakokinetiken för LEMTRADA har inte studerats hos patienter i åldern 55 år och äldre.
5.3 Prekliniska säkerhetsuppgifter
Karcinogenes och mutagenes
Inga studier har utförts för att bedöma den karcinogena eller mutagena potentialen för alemtuzumab.
Fertilitet och reproduktion
Behandling med intravenöst alemtuzumab med doser upp till 10 mg/kg/dag, administrerat under fem på varandra följande dagar (AUC 7,1 gånger den humana exponeringen vid rekommenderad daglig dos) hade ingen effekt på fertilitet och reproduktionsförmåga hos huCD52-transgena hanmöss. Antalet normala spermier var signifikant reducerat (<10%) jämfört med kontrollerna, och procentandelen onormala spermier (lossnade huvuden eller inga huvuden) hade ökat signifikant (upp till 3%). Dessa förändringar påverkade emellertid inte fertiliteten och ansågs därför inte allvarliga.
Hos honmöss som fick doser av alemtuzumab upp till 10 mg/kg/dag intravenöst (AUC 4,7 gånger den humana exponeringen vid rekommenderad daglig dos) under fem på varandra följande dagar före samboende med hanmöss av vild typ var det genomsnittliga antalet corpora lutea och implantationsställen per mus avsevärt reducerat jämfört med vehikelbehandlade djur. Minskad viktuppgång under dräktigheten i förhållande till vehikelkontrollerna observerades hos dräktiga möss som fått dosen 10 mg/kg/dag.
En reproduktionstoxicitetsstudie på dräktiga honmöss som fick doser av alemtuzumab upp till 10 mg/kg/dag intravenöst (AUC 2,4 gånger den humana exponeringen vid rekommenderad daglig dos 12 mg/dag) under fem på varandra följande dagar under dräktigheten resulterade i betydande ökningar av antalet kullar med samtliga foster döda eller resorberade, tillsammans med en åtföljande minskning av antalet kullar med livskraftiga foster. Inga externa, mjukvävnads- eller skelettmissbildningar eller variationer observerades vid doser upp till 10 mg/kg/dag.
Överföring till placenta och potentiell farmakologisk aktivitet av alemtuzumab observerades hos möss under dräktighet och efter födseln. I studier på möss observerades förändringar av antalet lymfocyter hos avkomma som exponerats för alemtuzumab under dräktigheten med doser upp till 3 mg/kg/dag, administrerat under fem på varandra följande dagar (AUC 0,6 gånger den humana exponeringen vid rekommenderad daglig dos 12 mg/dag). Kognitiv, fysisk och sexuell utveckling hos avkomma som exponerats för alemtuzumab under diandet påverkades inte vid doser upp till 10 mg/kg/dag av alemtuzumab.
6. FARMACEUTISKA UPPGIFTER
6.1 Förteckning över hjälpämnen
Dinatriumfosfatdihydrat (E339) Dinatriumedetatdihydrat Kaliumklorid (E508)
Kaliumdivätefosfat (E340)
Polysorbat 80 (E433)
Natriumklorid
Vatten för injektionsvätskor
6.2 Inkompatibiliteter
Då blandbarhetsstudier saknas får detta läkemedel inte blandas med andra läkemedel än de som omnämns i avsnitt 6.6.
6.3 Hållbarhet
Koncentrat 3 år
Utspädd lösning
Kemisk och fysiskalisk hållbarhet under användning har påvisats i 8 timmar vid 2°C - 8°C.
Ur mikrobiologisk synvinkel rekommenderas att produkten används omedelbart. Om den inte används omedelbart är förvaringstid och förvaringsförhållanden före användning användarens ansvar och skall normalt inte överstiga 8 timmar vid 2°C - 8°C, om den skyddas mot ljus.
6.4 Särskilda förvaringsanvisningar
Koncentrat.
Förvaras i kylskåp (2°C - 8°C).
Får ej frysas.
Förvara injektionsflaskan i ytterkartongen. Ljuskänsligt.
Förvaringsanvisningar för läkemedlet efter spädning finns i avsnitt 6.3.
6.5 Förpackningstyp och innehåll
LEMTRADA levereras i en 2 ml klar injektionsflaska med butylgummipropp och aluminiumförslutning med ett avsnäppbart lock av plast.
Förpackningsstorlek: kartong med en injektionsflaska.
6.6 Särskilda anvisningar för destruktion och övrig hantering
Innehållet i injektionsflaskan ska inspekteras för förekomst av partiklar och missfärgning före administrering. Använd den inte om det förekommer partiklar eller om koncentratet är missfärgat.
Skaka inte injektionsflaskorna före användning.
För intravenös administrering ska 1,2 ml LEMTRADA dras upp från injektionsflaskan till en spruta med aseptisk teknik. Injicera detta i 100 ml natriumklorid 9 mg/ml (0,9 %) infusionsvätska, lösning, eller glukos 50 mg/ml (5 %) infusionsvätska, lösning. Detta läkemedel får inte spädas med andra lösningsmedel. Påsen ska vändas försiktigt för att blanda lösningen.
LEMTRADA innehåller inga antimikrobiella konserveringsmedel och försiktighet ska därför iakttas för att säkerställa steriliteten hos den färdigberedda lösningen. Det rekommenderas att den utspädda produkten administreras omedelbart. Varje injektionsflaska är endast avsedd för engångsbruk.
Ej använt läkemedel och avfall ska kasseras enligt gällande anvisningar.
7. INNEHAVARE AV GODKÄNNANDE FÖR FÖRSÄLJNING
Genzyme Therapeutics Ltd 4620 Kingsgate Cascade Way
Oxford Business Park South Oxford OX4 2SU Storbritannien
8. NUMMER PÅ GODKÄNNANDE FÖR FÖRSÄLJNING
EU/1/13/869/001
9. DATUM FÖR FÖRSTA GODKÄNNANDE/FÖRNYAT GODKÄNNANDE
Datum för det första godkännandet: 12 september 2013
10. DATUM FÖR ÖVERSYN AV PRODUKTRESUMÉN
Ytterligare information om detta läkemedel finns på Europeiska läkemedelsmyndighetens webbplats http://www.ema.europa.eu.
A. TILLVERKAREN AV DEN AKTIVA SUBSTANSEN AV BIOLOGISKT URSPRUNG OCH TILLVERKARE SOM ANSVARAR FÖR FRISLÄPPANDE AV TILLVERKNINGSSATS
B. VILLKOR ELLER BEGRÄNSNINGAR FÖR TILLHANDAHÅLLANDE OCH ANVÄNDNING
C. ÖVRIGA VILLKOR OCH KRAV FÖR GODKÄNNANDET FÖR FÖRSÄLJNING
D. VILLKOR ELLER BEGRÄNSNINGAR AVSEENDE EN SÄKER OCH EFFEKTIV ANVÄNDNING AV LÄKEMEDLET
A. TILLVERKAREN AV DEN AKTIVA SUBSTANSEN AV BIOLOGISKT URSPRUNG OCH TILLVERKARE SOM ANSVARAR FÖR FRISLÄPPANDE AV TILLVERKNINGSSATS
Namn och adress till tillverkare av aktiv substans av biologiskt ursprung
Boehringer Ingelheim Pharma GmbH & Co. KG Birkendorfer Straße 65 88397 Biberach an der Riss TYSKLAND
Namn och adress till tillverkare som ansvarar för frisläppande av tillverkningssats
Genzyme Limited
37 Hollands Road
Haverhill
Suffolk
CB9 8PU
Storbritannien
Genzyme Ireland Limited IDA Industrial Park Old Kilmeaden Road Waterford Irland
I läkemedlets tryckta bipacksedel ska namn och adress till tillverkaren som ansvarar för frisläppandet av den relevanta tillverkningssatsen anges.
B. VILLKOR ELLER BEGRÄNSNINGAR FÖR TILLHANDAHÅLLANDE OCH ANVÄNDNING
Läkemedel som med begränsningar lämnas ut mot recept (se bilaga I: Produktresumén, avsnitt 4.2).
C. ÖVRIGA VILLKOR OCH KRAV FÖR GODKÄNNANDET FÖR FÖRSÄLJNING
• Periodiska säkerhetsrapporter
Innehavaren av godkännandet för försäljning ska lämna in den första periodiska säkerhetsrapporten för detta läkemedel inom 6 månader efter godkännandet. Sedan ska innehavaren av godkännandet för försäljning lämna in periodiska säkerhetsrapporter för detta läkemedel i enlighet med de krav som anges i den förteckning över referensdatum för unionen (EURD-listan) som föreskrivs i artikel 107c.7 i direktiv 2001/83/EG och som offentliggjorts på webbportalen för europeiska läkemedel.
D. VILLKOR ELLER BEGRÄNSNINGAR AVSEENDE EN SÄKER OCH EFFEKTIV ANVÄNDNING AV LÄKEMEDLET
• Riskhanteringsplan
Innehavaren av godkännandet för försäljning ska genomföra de erforderliga farmakovigilansaktiviteter och -åtgärder som finns beskrivna i den överenskomna riskhanteringsplanen (Risk Management Plan, RMP) som finns i modul 1.8.2. i godkännandet för försäljning samt eventuella efterföljande överenskomna uppdateringar av riskhanteringsplanen.
En uppdaterad riskhanteringsplan ska lämnas in
• på begäran av Europeiska läkemedelsmyndigheten,
• när riskhanteringssystemet ändras, särskilt efter att ny information framkommit som kan leda till
betydande ändringar i läkemedlets nytta-riskprofil eller efter att en viktig milstolpe (för farmakovigilans eller riskminimering) har nåtts.
Om datum för inlämnandet av en periodisk säkerhetsrapport och uppdateringen av en riskhanteringsplan sammanfaller kan de lämnas in samtidigt.
• Ytterligare riskminimeringsåtgärder
Före lansering ska innehavaren av godkännandet för försäljning ha överenskommit med den nationella behöriga myndigheten i varje medlemsstat om ett utbildningsprogram för hälso- och sjukvårdspersonal samt patienter.
Enligt överenskommelse med den nationella behöriga myndigheten i varje medlemsstat där LEMTRADA marknadsförs ska innehavaren av godkännandet för försäljning säkerställa, både vid lansering och efter lansering, att alla läkare som avser att förskriva LEMTRADA får ett uppdaterat utbildningspaket avsett för läkare med följande innehåll:
• produktresumé
• guide för hälso- och sjukvårdspersonal
• checklista för förskrivare
• patientguide
• patientvarningskort
Guiden för hälso- och sjukvårdspersonal ska innehålla följande huvudbudskap:
1. En beskrivning av riskerna associerade med användning av LEMTRADA, dvs. :
• immunologisk trombocytopeni (ITP)
• nefropatier, inklusive anti-glomerulär basalmembranssjukdom (anti-GBM)
• tyreoidearubbningar
2. Rekommendationer om hur dessa risker kan minskas genom lämplig patientrådgivning, -övervakning och -hantering.
3. Ett avsnitt med vanliga frågor och svar
Checklistan för förskrivare ska innehålla följande huvudbudskap:
1. Listor över provtagningar som ska utföras för initial screening av patienten.
2. Vaccinationsprogram som ska genomföras 6 veckor före behandling.
3. Premedicinering, allmänt hälsotillstånd, graviditet och preventivmedel ska kontrolleras omedelbart före behandling.
4. Monitoreringsaktiviteter under behandling och under 4 år efter den sista behandlingen.
5. En specifik referens till det faktum att patienten har informerats och förstår riskerna av allvarliga autoimmuna sjukdomar, infektioner och maligniteter samt försiktighetsåtgärderna för att minimera dessa risker.
Patientguiden ska innehålla följande huvudbudskap:
1. En beskrivning av riskerna associerade med användning av LEMTRADA, dvs.:
• immunologisk trombocytopeni (ITP)
• nefropatier, inklusive anti-glomerulär basalmembranssjukdom (anti-GBM)
• sköldkörtelrubbningar
• allvarliga infektioner
2. En beskrivning av tecken och symtom på autoimmuna risker.
3. En beskrivning av den bästa åtgärden om tecken och symtom på dessa risker upptäcks (t.ex. kontaktuppgifter till din läkare).
4. Rekommendationer för planering av uppföljningsschema.
Patientvarningskortet ska innehålla följande huvudbudskap:
1. Ett varningsmeddelande för hälso- och sjukvårdspersonal som behandlar patienten, inklusive vid nödsituationer, om att patienten har behandlats med LEMTRADA.
2. Att behandling med LEMTRADA kan öka risken för:
• immunologisk trombocytopeni (ITP)
• nefropatier, inklusive anti-glomerulär basalmembranssjukdom (anti-GBM)
• tyreoidearubbningar
• allvarliga infektioner
3. Kontaktuppgifter till förskrivaren av LEMTRADA.
MÄRKNING OCH BIPACKSEDEL
A. MÄRKNING
LEMTRADA 12 mg koncentrat till infusionsvätska, lösning alemtuzumab
Varje injektionsflaska innehåller 12 mg alemtuzumab i 1,2 ml (10 mg/ml).
E339, dinatriumedetatdihydrat, E508, E340, E433, natriumklorid, vatten för injektionsvätskor
Koncentrat till infusionsvätska, lösning 1 injektionsflaska 12 mg/1,2 ml
Läs bipacksedeln före användning. Intravenös användning.
Administreras inom 8 timmar efter spädning.
Förvaras utom syn- och räckhåll för barn.
EXP
Förvara injektionsflaskan i ytterkartongen. Ljuskänsligt. Förvaras i kylskåp.
Får ej frysas-eller skakas.
10. SÄRSKILDA FÖRSIKTIGHETSÅTGÄRDER FÖR DESTRUKTION AV EJ ANVÄNT LÄKEMEDEL OCH AVFALL I FÖREKOMMANDE FALL
11. INNEHAVARE AV GODKÄNNANDE FÖR FÖRSÄLJNING (NAMN OCH ADRESS)
Genzyme Therapeutics Ltd 4620 Kingsgate Cascade Way
Oxford Business Park South Oxford OX4 2SU Storbritannien
12. NUMMER PÅ GODKÄNNANDE FÖR FÖRSÄLJNING
EU/1/13/869/001
13. TILLVERKNINGSSATSNUMMER
Lot
14. ALLMÄN KLASSIFICERING FÖR FÖRSKRIVNING
15. BRUKSANVISNING
16. INFORMATION I PUNKTSKRIFT
Braille krävs ej.
1. LÄKEMEDLETS NAMN OCH ADMINISTRERINGSVÄG
LEMTRADA 12 mg sterilt koncentrat
alemtuzumab
i.v.
2. ADMINISTRERINGSSÄTT
3. UTGÅNGSDATUM
EXP
4. TILLVERKNINGSSATSNUMMER
Lot
5. MÄNGD UTTRYCKT I VIKT, VOLYM ELLER PER ENHET
1,2 ml
6. ÖVRIGT
B. BIPACKSEDEL
Bipacksedel: Information till patienten
LEMTRADA 12 mg koncentrat till infusionsvätska, lösning
alemtuzumab
Detta läkemedel är föremål för utökad övervakning. Detta kommer att göra det möjligt att snabbt identifiera ny säkerhetsinformation. Du kan hjälpa till genom att rapportera de biverkningar du eventuellt får. Information om hur du rapporterar biverkningar finns i slutet av avsnitt 4.
Läs noga igenom denna bipacksedel innan du får detta läkemedel. Den innehåller information som är viktig för dig.
- Spara denna information, du kan behöva läsa den igen.
- Om du har ytterligare frågor vänd dig till läkare.
- Om du får biverkningar, tala med läkare. Detta gäller även eventuella biverkningar som inte nämns i denna information. Se avsnitt 4.
I denna bipacksedel finns information om följande:
1. Vad LEMTRADA är och vad det används för
2. Vad du behöver veta innan du får LEMTRADA
3. Hur du får LEMTRADA
4. Eventuella biverkningar
5. Hur LEMTRADA ska förvaras
6. Förpackningens innehåll och övriga upplysningar
1. Vad LEMTRADA är och vad det används för
LEMTRADA innehåller den aktiva substansen alemtuzumab, som används för att behandla en form av multipel skleros (MS) hos vuxna, som kallas skovvis förlöpande MS (RRMS, relapsing remitting MS). LEMTRADA botar inte MS, men kan reducera antalet skov av MS. LEMTRADA kan också bidra till att vissa tecken och symtom på MS bromsas eller går tillbaka. I kliniska prövningar fick patienter som behandlades med LEMTRADA färre skov och var mindre benägna att få försämrad funktionsnedsättning jämfört med patienter som behandlades med en beta-interferon som injicerades flera gånger i veckan.
Vad är multipel skleros?
Multipel skleros, MS, är en autoimmun sjukdom som drabbar det centrala nervsystemet (hjärnan och ryggmärgen). MS innebär att ditt immunsystem av misstag går till angrepp på det skyddande lagret (myelin) runt nervtrådarna och orsakar inflammation. När inflammationen orsakar symtom kallas det ofta för en attack eller ett skov. Patienter med RRMS upplever skov som följs av perioder av tillfrisknande.
Vilka symtom du upplever beror på vilken del av ditt centrala nervsystem som är drabbat. Den skada som uppstår på dina nerver under den här inflammationen kan gå tillbaka, men efterhand som sjukdomen framskrider kan skadan byggas på och bli permanent.
Hur LEMTRADA fungerar
LEMTRADA reglerar ditt immunsystem för att begränsa angreppen på nervsystemet.
2. Vad du behöver veta innan du får LEMTRADA Använd INTE LEMTRADA:
- om du är allergisk mot alemtuzumab eller något annat innehållsämne i detta läkemedel (anges i avsnitt 6)
- om du är infekterad med humant immunbristvirus (hiv).
Varningar och försiktighet
Tala med din läkare innan du får LEMTRADA. Efter att du har fått en behandlingskur med LEMTRADA kan du löpa större risk för att utveckla andra autoimmuna tillstånd eller allvarliga infektioner. Det är viktigt att du är införstådd med dessa risker och hur du ska hålla dem under uppsikt. Du kommer att få ett patientvarningskort och en patientguide med ytterligare information. Det är viktigt att du alltid har patientvarningskortet med dig under behandlingen och under fyra år efter din sista infusion med LEMTRADA, eftersom biverkningar kan uppstå flera år efter behandlingen. Du ska alltid visa patientvarningskortet för läkaren när du får medicinsk behandling, även om det inte är för din MS.
Läkaren kommer att ta blodprover innan du påbörjar behandlingen med LEMTRADA. Syftet med proverna är att undersöka om du kan ta LEMTRADA. Läkaren vill också vara säker på att du inte har vissa medicinska tillstånd eller sjukdomar innan du påbörjar behandlingen med LEMTRADA.
• Autoimmuna tillstånd
Behandling med LEMTRADA kan öka risken för autoimmuna tillstånd. Sådana tillstånd innebär att immunsystemet av misstag angriper din kropp. Nedan hittar du information om vissa specifika tillstånd som har observerats hos MS-patienter som har behandlats med LEMTRADA.
Dessa autoimmuna tillstånd kan uppstå flera år efter behandling med LEMTRADA. Därför måste du lämna regelbundna blod- och urinprover i fyra år efter din sista infusion. Dessa prover behövs även om du mår bra och om dina MS-symtom är under kontroll. Dessutom finns det vissa tecken och symtom som du själv bör hålla utkik efter. Detaljerad information om dessa tecken och symtom, provtagningen och åtgärder som du behöver vidta finns i avsnitt 4 - autoimmuna tillstånd.
Ytterligare användbar information om dessa autoimmuna tillstånd och hur man testar för dem finns i patientguiden för LEMTRADA.
o Immunologisk trombocytopeni (ITP)
En mindre vanlig biverkan är att patienter utvecklat en blödningssjukdom som orsakas av en låg nivå av blodplättar och kallas immunologisk trombocytopeni (ITP). Den här sjukdomen måste diagnostiseras och behandlas tidigt, annars kan effekterna bli allvarliga eller till och med dödliga. En beskrivning av tecken och symtom på ITP finns i avsnitt 4.
0 Njursjukdom (till exempel anti-GBM-sjukdom)
1 sällsynta fall har patienter upplevt autoimmuna reaktioner som har orsakat problem med njurarna, till exempel antiglomerulär basalmembransjukdom (anti-GBM-sjukdom). En beskrivning av tecken och symtom på njursjukdom finns i avsnitt 4. Om den inte behandlas kan den orsaka njursvikt som kräver dialys eller transplantation, och kan leda till döden.
o Sköldkörtelrubbningar
Det är mycket vanligt att patienter har upplevt en autoimmun rubbning av sköldkörteln som påverkar dess förmåga att bilda eller kontrollera hormoner som är viktiga för ämnesomsättningen.
LEMTRADA kan orsaka olika typer av sköldkörtelrubbningar, däribland:
• Överaktiv sköldkörtel (hypertyreos), där sköldkörteln producerar för mycket hormon
• Underaktiv sköldkörtel (hypotyreos), där sköldkörteln inte producerar tillräckligt med hormon.
En beskrivning av tecken och symtom på sköldkörtelrubbningar finns i avsnitt 4.
Om du utvecklar en sköldkörtelrubbning måste du i de flesta fall behandlas resten av livet med läkemedel som kontrollerar sköldkörtelrubbningen, och i vissa fall kan sköldkörteln behöva tas bort.
Det är mycket viktigt att du får rätt behandling vid sköldkörtelrubbning, särskilt om du blir gravid efter att ha använt LEMTRADA. Om du har en obehandlad sköldkörtelrubbning kan detta skada fostret eller det nyfödda barnet.
0 Andra autoimmuna tillstånd
1 sällsynta fall har patienter upplevt autoimmuna tillstånd som involverar röda eller vita blodkroppar. Detta kan diagnostiseras från de blodprover som du måste lämna regelbundet efter behandlingen med LEMTRADA. Om du utvecklar något av dessa tillstånd kommer läkaren att berätta det för dig och vidta lämpliga åtgärder för att behandla tillståndet.
• Infusionsreaktioner
De flesta patienter som behandlas med LEMTRADA upplever biverkningar under själva infusionen eller inom 24 timmar efter infusionen. För att försöka minska infusionsreaktionerna kommer läkaren att ge dig något eller några andra läkemedel (se avsnitt 4 - infusionsreaktioner).
• Infektioner
Patienter som behandlas med LEMTRADA löper högre risk att drabbas av en allvarlig infektion (se avsnitt 4 - infektioner). I allmänhet kan dessa infektioner behandlas med standardläkemedel.
För att minska risken för att du ska få en infektion kommer läkaren att kontrollera om andra läkemedel som du tar kan påverka ditt immunsystem. Därför är det viktigt att du berättar för läkaren om alla läkemedel som du tar.
Om du har en infektion innan du påbörjar behandlingen med LEMTRADA kommer läkaren att överväga att skjuta upp behandlingen tills infektionen är under kontroll är eller har gått över.
Patienter som behandlas med LEMTRADA löper högre risk att utveckla herpesinfektion (till exempel ett munsår). I allmänhet har en patient som redan har haft en herpesinfektion högre risk att utveckla en herpesinfektion på nytt. Det är även möjligt att utveckla en herpesinfektion för första gången. Det är rekommenderat att din läkare skriver ut ett läkemedel som minskar risken att utveckla en herpesinfektion som du ska ta de dagar då du får behandling med LEMTRADA och i en månad efter behandlingen.
Dessutom finns risken att utveckla infektioner som kan leda till förändringar i livmoderhalsen (cervix). Därför är det rekommenderat att alla kvinnliga patienter genomgår en årlig undersökning, såsom cellprov. Läkaren förklarar vilka tester som behövs.
Patienter som behandlas med LEMTRADA löper också en högre risk att drabbas av listerios eller listeria-meningit (hjärnhinneinflammation). För att minska denna risk ska du undvika att äta rått eller dåligt tillagat kött, möge- och kittost och opastöriserade mejeriprodukter i minst en månad efter behandling med LEMTRADA.
Om du bor i ett område där tuberkulosinfektioner är vanliga kan du löpa större risk att drabbas av infektion med tuberkulos. Din läkare ordnar med kontroller för tuberkulos.
Om du är bärare av hepatit B- eller hepatit C-infektion (dessa sjukdomar påverkar levern) måste extra försiktighet iakttas innan du kan få behandling med LEMTRADA, eftersom det är okänt om behandlingen skulle kunna leda till att hepatitinfektionen aktiveras, något som i så fall kan skada din lever.
• Tidigare cancerdiagnos
Informera din läkare om du tidigare har diagnostiserats med cancer.
• Vaccin
Det är okänt om LEMTRADA påverkar ditt svar på vaccin. Om du inte har genomgått de vanliga standardvaccinationerna kommer din läkare att överväga om du bör få dem innan du får behandling med LEMTRADA. I synnerhet kommer läkaren att överväga att vaccinera dig mot vattkoppor om du aldrig har haft det. Eventuella vaccinationer måste göras minst sex veckor innan du påbörjar en behandlingskur med LEMTRADA.
Du får INTE ta vissa typer av vacciner (vacciner med levande virus) om du nyligen har fått LEMTRADA.
Barn och ungdomar
LEMTRADA är inte avsett att användas till barn och ungdomar under 18 år eftersom det inte har studerats hos MS-patienter under 18 års ålder.
Andra läkemedel och LEMTRADA
Tala om för läkare eller apotekspersonal om du tar, nyligen har tagit eller kan tänkas ta andra läkemedel (inklusive eventuella vaccinationer eller naturläkemedel).
Förutom LEMTRADA finns det andra behandlingar, både mot MS och för behandling av andra tillstånd, som kan påverka ditt immunsystem och därför påverka din förmåga att bekämpa infektioner. Om du använder något sådant läkemedel kan läkaren be dig att sluta ta det läkemedlet innan du påbörjar behandlingen med LEMTRADA.
Graviditet
Om du är gravid, tror att du kan vara gravid eller planerar att skaffa barn, rådfråga läkare innan du får detta läkemedel.
Fertila kvinnor ska använda effektiv preventivmetod under varje behandlingskur med LEMTRADA och under fyra månader efter varje behandlingskur.
Om du blir gravid efter behandling med LEMTRADA och drabbas av en sköldkörtelrubbning under graviditeten krävs extra försiktighet. Sköldkörtelrubbningar kan skada barnet (se avsnitt 2 Varningar och försiktighet - autoimmuna tillstånd).
Amning
Det är inte känt om LEMTRADA kan överföras till spädbarn via bröstmjölk, men det finns en risk att det kan vara så. Därför är det rekommenderat att du inte ammar under behandlingskurerna med LEMTRADA och i fyra månader efter varje behandlingskur. Samtidigt kan det finnas fördelar med bröstmjölk som kan bidra till att skydda barnet från infektioner. Därför ska du tala med din läkare om du planerar att amma ditt barn för att få råd om vad som är bäst för dig och ditt barn.
Fertilitet
Under behandlingskuren och i fyra månader efteråt kan LEMTRADA finnas kvar i din kropp. Det är inte känt om LEMTRADA har någon effekt på fertiliteten under denna period. Tala med läkaren om du planerar att bli gravid.
Körförmåga och användning av maskiner
Många patienter upplever biverkningar när de får infusionen eller inom 24 timmar efter infusionen med LEMTRADA, och vissa av dessa, till exempel yrsel, kan göra det osäkert att framföra fordon eller använda maskiner. Om du drabbas av detta ska du avbryta dessa aktiviteter tills du mår bättre.
LEMTRADA innehåller kalium och natrium
Det här läkemedlet innehåller mindre än 1 mmol kalium (39 mg) per infusion, d.v.s. är näst intill ”kaliumfritt”.
Det här läkemedlet innehåller mindre än 1 mmol natrium (23 mg) per infusion, d.v.s. är näst intill ”natriumfritt”.
3. Hur du får LEMTRADA
Läkaren kommer att förklara för dig hur det går till när du får LEMTRADA. Om du har frågor vänd dig till läkaren.
Under den första behandlingskuren kommer du att få en infusion per dag i fem dagar (kur 1).
Ett år senare kommer du att få en infusion per dag i tre dagar (kur 2).
Du får ingen behandling med LEMTRADA mellan dessa två kurer.
Maximal dagsdos är en infusion.
Du kommer att få LEMTRADA som en infusion i en ven. Varje infusion tar ungefär fyra timmar. För de flesta patienter gör två behandlingskurer att MS-aktiviteten reduceras i två år. Övervakning med avseende på biverkningar och regelbunden provtagning måste fortsätta i fyra år efter den sista infusionen.
Diagrammet nedan kan hjälpa dig att förstå hur länge effekten av behandlingen varar och hur lång uppföljning som krävs.
Inled blod- oh urinprover före behandling
-► och fortsätt i 4 år efter den sista infusionen
Behandlingskur 1 Behandlingskur 2
lllll III
5 dagars behandling 3 dagars behandling
År 1 År 2 År 3 År 4 År 5
OBS För de flesta patienter verkar två behandlingskurer i två år eller längre.
|
BEHANDLING | |||
|
UPPFÖLJNING | |||
Uppföljning efter behandling med LEMTRADA
När du har fått LEMTRADA måste du genomgå regelbundna tester för att säkerställa att eventuella biverkningar kan diagnostiseras och behandlas snarast. Testerna måste fortsätta i fyra år efter den sista infusionen. Se beskrivning i avsnitt 4 - de viktigaste biverkningarna.
Om du får för stor mängd av LEMTRADA
Patienter som av misstag har fått för stor mängd LEMTRADA i en infusion har upplevt allvarliga reaktioner som huvudvärk, utslag, lågt blodtryck eller ökad hjärtfrekvens. Doser som är högre än den rekommenderade dosen kan leda till allvarligare eller mer långvariga infusionsreaktioner (se avsnitt 4) eller en mer kraftfull effekt på immunsystemet. Vid överdosering avbryter man administreringen av LEMTRADA och behandlar symtomen.
Om du har ytterligare frågor om detta läkemedel, kontakta läkare.
4. Eventuella biverkningar
Liksom alla läkemedel kan detta läkemedel orsaka biverkningar, men alla användare behöver inte få dem.
De viktigaste biverkningarna är de autoimmuna tillstånd som beskrivs i avsnitt 2, däribland:
• ITP (blödningssjukdom) (mindre vanlig - kan förekomma hos upp till 1 av 100 användare): kan visa sig som små utströdda röda, rosa eller lila prickar på huden; att man lättare får blåmärken; att blödningar från sår är svårare att stoppa; kraftigare, mer långvariga eller frekventa mensblödningar än vanligt; blödningar mellan menstruationerna; blödning från tandkött eller näsa som har uppstått nyligen eller tar längre tid än vanligt att stoppa; eller att man hostar upp blod.
• njursjukdom (sällsynt - kan förekomma hos upp till 1 av 1000 användare): kan visa sig som blod i urinen (din urin kan vara röd eller ha samma färg som te) eller som svullnad i ben eller fötter. Det kan också leda till skada på lungorna, vilket kan resultera i att du hostar upp blod
Om du upplever några av dessa tecken eller symtom på blödnings- eller njursjukdom ska du omedelbart kontakta läkaren och berätta om symtomen. Om du inte kan få tag i din vanliga läkare måste du omedelbart söka vård på annat håll._
• sköldkörtelrubbningar (mycket vanlig - kan förekomma hos fler än 1 av 10 användare): kan visa sig som överdrivna svettningar; oförklarlig viktnedgång eller viktuppgång; ögonsvullnad; nervositet; snabb hjärtrytm; att man fryser; trötthet som blir värre eller nyligen uppkommen förstoppning.
• rubbningar av röda eller vita blodkroppar (sällsynt - kan förekomma hos upp till 1 av 1000 användare): som konstateras med hjälp av blodprov.
Du kommer att få lämna följande prover med avseende på autoimmuna tillstånd:
|
Prov |
När? |
Hur länge? |
|
Blodprov (för att kunna diagnostisera alla viktiga allvarliga biverkningar som anges ovan) |
Innan behandlingen inleds och varje månad efter behandlingen |
Till och med fyra år efter din sista LEMTRADA-infusion |
|
Urinprov (ytterligare prov för att diagnostisera njursjukdomar) |
Innan behandlingen inleds och varje månad efter behandling |
Till och med fyra år efter din sista LEMTRADA-infusion |
Alla dessa allvarliga biverkningar kan uppkomma flera år efter att du har fått LEMTRADA. Om du upplever några av dessa tecken eller symtom ska du omedelbart kontakta läkaren och berätta om dem. Du kommer också att få lämna blod- och urinprov regelbundet för att du snabbt ska kunna få behandling om du skulle drabbas av något av dessa tillstånd._
Efter denna period kommer läkaren att utföra fler provtagningar om du får symtom på ITP, njur- eller sköldkörtelrubbningar. Efter de fyra åren ska du fortsätta vara uppmärksam på tecken eller symtom på biverkningar så som beskrivs i din patientguide, och du ska fortsätta bära med dig patientvarningskortet.
En annan viktig biverkning är den ökade risken för infektioner (nedan finns information om hur ofta patienter drabbas av infektioner). Dessa är i regel lindriga, men allvarliga infektioner kan förekomma.
Tala omedelbart om för läkaren om du upplever några av dessa tecken på infektion
• feber och/eller frossa
• svullna körtlar
För att minska risken för vissa infektioner kan läkaren överväga att ge dig en vaccination mot vattkoppor och/eller andra vaccinationer som läkaren anser är nödvändiga för dig (se avsnitt 2: Vad du behöver veta innan du får LEMTRADA - Vaccin). Läkaren kan också skriva ut läkemedel mot munsår (se avsnitt 2: Vad du behöver veta innan du får LEMTRADA - Infektioner).
De vanligaste biverkningarna är infusionsreaktioner (nedan finns information om hur ofta patienterna upplever sådana), som kan uppstå under själva infusionen eller inom 24 timmar efter infusionen. Dessa är i regel lindriga men allvarliga reaktioner kan förekomma. Ibland kan även allergiska reaktioner förekomma.
För att försöka minska infusionsreaktionerna kommer läkaren att ge dig andra läkemedel (kortikosteroider) före de tre första infusionerna i varje behandlingskur med LEMTRADA. Det finns även andra behandlingar som kan begränsa dessa reaktioner och som du kan få före infusionen eller när du upplever symtom. Dessutom kommer du att övervakas under infusionen och i två timmar efter att infusionen har avslutats. Om du skulle få allvarliga reaktioner kan man sänka hastigheten på infusionen eller avbryta den helt.
Mer information om dessa reaktioner hittar du i patientguiden för LEMTRADA.
De biverkningar som du kan komma att uppleva är följande:
Mycket vanliga (kan förekomma hos fler än 1 av 10 användare):
• Infusionsreaktioner som kan inträffa under själva infusionen eller inom 24 timmar efter infusionen: huvudvärk, utslag, feber, illamående, nässelfeber, klåda, rodnad i ansiktet och på halsen, trötthetskänsla
• Infektioner: luftvägsinfektioner som förkylning och bihåleinflammation, urinvägsinfektion
• Minskat antal vita blodkroppar (lymfocyter)
Vanliga (kan förekomma hos upp till 1 av 10 användare):
• Infusionsreaktioner som kan inträffa under själva infusionen eller inom 24 timmar efter infusionen: förändringar i hjärtfrekvens, matsmältningsbesvär, frossa, obehag i bröstet, smärta, yrsel, smakförändringar, sömnsvårigheter, andningssvårigheter eller andfåddhet, utslag på kroppen, lågt blodtryck.
• Infektioner: hosta, öroninflammation, influensaliknande sjukdom, bronkit, lunginflammation, oral eller vaginal svamp, bältros, vattkoppor, munsår, svullna eller förstorade körtlar
• smärta vid infektionsstället, smärta i ryggen, nacken, i armar eller ben, muskelsmärtor, muskelkramper, ledvärk, smärta i munnen eller halsen
• inflammation i munnen/tandköttet/tungan
• allmänt obehag, svaghet, kräkningar, diarré, magsmärtor, maginfluensa
• halsbränna
• avvikelser som kan visa sig vid undersökningar: blod eller protein i urinen, minskad hjärtfrekvens, oregelbunden eller onormal hjärtrytm, högt blodtryck
• MS-skov
• skakningar, känselbortfall, brännande eller stickande känsla
• överaktiv eller underaktiv sköldkörtel, eller struma (förstoring av sköldkörteln, som sitter på halsen)
• svullna armar och/eller ben
• synproblem
• oroskänsla
• ovanligt kraftiga, långvariga eller oregelbundna menstruationsblödningar
• akne, hudrodnad, överdriven svettning
• näsblod, blåmärken
• håravfall
Mindre vanliga (kan förekomma hos upp till 1 av 100 användare):
• Infektioner: könsherpes, ögoninfektion, tandinfektion
• problem med blodkoagulationen, blodbrist (anemi)
• fotsvamp
• avvikande vaginalt cellprov
• depression
• ökad känsel
• svårighet att svälja
• hicka
• viktminskning
• förstoppning
• blödning från tandköttet
• avvikande leverprover
• blåsor
Visa patientvarningskortet och den här bipacksedeln för alla läkare som är involverade i din behandling, inte bara för neurologen.
Den här informationen finns även på patientvarningskortet och i patientguiden som du har fått av läkaren.
Rapportering av biverkningar
Om du får biverkningar, tala med läkare. Detta gäller även biverkningar som inte nämns i denna information. Du kan också rapportera biverkningar direkt via det nationella rapporteringssystemet listat i bilaga V. Genom att rapportera biverkningar kan du bidra till att öka informationen om läkemedels säkerhet.
5. Hur LEMTRADA ska förvaras
Förvara detta läkemedel utom syn- och räckhåll för bam.
Används före utgångsdatum som anges på ytterkartongen och injektionsflaskan efter EXP. Utgångsdatumet är den sista dagen i angiven månad.
Förvaras i kylskåp (2 °C-8 °C).
Får ej frysas.
Förvaras i originalförpackningen. Ljuskänsligt.
Det är rekommenderat att läkemedlet används omedelbart efter spädning, på grund av möjlig risk för mikrobiell kontaminering. Om läkemedlet inte används omedelbart är det användarens ansvar att se till att förvaringstiden under användning samt förhållandena före användning inte överstiger 8 timmar vid 2 °C till 8 °C, skyddat från ljus.
Använd inte detta läkemedel om det finns partiklar i vätskan och/eller om vätskan i injektionsflaskan är missfärgad.
Läkemedel ska inte kastas i avloppet. Dessa åtgärder är till för att skydda miljön.
6. Förpackningens innehåll och övriga upplysningar
Innehållsdeklaration
Den aktiva substansen är alemtuzumab.
Varje injektionsflaska innehåller 12 mg alemtuzumab i 1,2 ml.
Övriga innehållsämnen är:
• dinatriumfosfatdihydrat (E339)
• dinatriumedetatdihydrat
• kaliumklorid (E508)
• kaliumdivätefosfat (E340)
• polysorbat 80 (E433)
• natriumklorid
• vatten till injektionsvätskor
Läkemedlets utseende och förpackningsstorlekar
LEMTRADA är ett klart, färglöst till svagt gulfärgat koncentrat till infusionsvätska lösning (sterilt koncentrat) som levereras i en injektionsflaska av glas med propp.
Det finns 1 injektionsflaska i varje kartong.
Innehavare av godkännande för försäljning
Genzyme Therapeutics Ltd, 4620 Kingsgate, Cascade Way, Oxford Business Park South, Oxford, OX4 2SU, Storbritannien.
Tillverkare
Genzyme Ltd., 37 Hollands Road, Haverhill, Suffolk CB9 8PU, Storbritannien.
Genzyme Ireland Limited, IDA Industrial Park, Old Kilmeaden Road, Waterford, Irland.
Kontakta ombudet för innehavaren av godkännandet för försäljning om du vill veta mer om detta läkemedel
|
Belgie/Belgique/Belgien/ Luxemburg/Luxembourg Sanofi Belgium Tél/Tel: + 32 2 710 54 00 |
Lietuva UAB „SANOFI-AVENTIS LIETUVA“ Tel. +370 5 275 5224 |
|
Etnrapua Sanofi-Aventis Bulgaria EOOD Ten: +359 2 9705300 |
Magyarorszag sanofi-aventis Zrt Tel: +36 1 505 0050 |
|
Ceská republika sanofi-aventis, s.r.o. Tel: +420 233086 111 |
Malta Sanofi-Aventis Malta Ltd Tel: +356 21493022 |
|
Danmark sanofi-aventis Denmark A/S Tlf: +45 45 16 70 00 |
Nederland Genzyme Europe B.V. Tel: +31 35 699 1200 |
|
Deutschland Genzyme Therapeutics Ltd. Tel: +49 (0) 6102 3674 451 |
Norge sanofi-aventis Norge AS Tlf: + 47 67 10 71 00 |
|
Eesti sanofi-aventis Estonia OÜ Tel. +372 6 273 488 |
Österreich sanofi-aventis GmbH Tel: + 43 1 80 185 - 0 |
|
Ekkáda/Kúnpo^ sanofi-aventis AEBE (ELLáSa) T r(k: +30 210 900 1600 |
Polska sanofi-aventis Sp. z o.o. Tel.: +48 22 280 00 00 |
|
España Genzyme, S.L.U. Tel: +34 93 485 94 00 sanofi-aventis, S.A. Tel: +34 93 485 94 00 |
Portugal Sanofi - Produtos Farmaceuticos, Lda. Tel: +351 21 422 0100 |
|
France Genzyme S.A.S. Tél : +33 (0) 825 825 863 |
Romania sanofi-aventis Romania S.R.L. Tel: +40 (0) 21 317 31 36 |
|
Hrvatska sanofi-aventis Croatia d.o.o. Tel: +385 1 6003 400 |
Slovenija sanofi-aventis d.o.o. Tel: +386 1 560 4800 |
Slovenska republika
Island
Vistor hf.
Simi: +354 535 7000
sanofi-aventis Pharma Slovakia s.r.o. Tel.: +421 2 33 100 100
Ireland Suomi/Finland
Genzyme Therapeutics Ltd. (United Kingdom) sanofi-aventis Oy
Tel: +44 (0) 1865 405200 Puh/Tel: + 358 201 200 300
Sverige
sanofi-aventis AB
Tel: +46 (0)8 634 50 00
Italia
Genzyme Srl Tel: +39 059 349 811
Latvija
sanofi-aventis Latvia SIA Tel: +371 67 33 24 51
United Kingdom
Genzyme Therapeutics Ltd. (United Kingdom)
Tel: +44 (0) 1865 405200
Denna bipacksedel ändrades senast Övriga informationskällor
Följande material finns tillgängliga för att minimera riskerna och förse patienterna med information om möjliga biverkningar och instruktioner om vad man bör göra om vissa biverkningar uppträder:
1. Patientvarningskort: Patienten ska uppvisa detta för andra vårdgivare för att upplysa dem om att
patienten använder LEMTRADA
2. Patientguide: För ytterligare information om autoimmuna reaktioner och infektioner samt övriga
upplysningar.
Ytterligare information om detta läkemedel finns på Europeiska läkemedelsmyndighetens webbplats: http://www.ema.europa.eu/.
Följande uppgifter är endast avsedda för hälso- och sjukvårdspersonal:
Information om riskminimering - autoimmuna tillstånd
• Det är av yttersta vikt att patienten förstår åtagandet att lämna regelbundna prover i fyra år efter den sista infusionen även om patienten är fri från symtom och MS-sjukdomen är under kontroll.
• Du och din patient måste tillsammans planera och sköta den regelbundna uppföljningen.
• Om patienten inte följer rekommendationerna kan det behövas ytterligare rådgivning för att lyfta fram riskerna med att missa planerade provtagningar.
• Övervaka provresultaten och var uppmärksam på symtom på biverkningar.
• Gå igenom patientguiden och bipacksedeln för LEMTRADA med din patient. Påminn patienten om att vara uppmärksam på symtom på autoimmuna tillstånd och att söka vård vid eventuell oro.
Följande utbildningsmaterial för vårdpersonal finns också tillgängligt:
LEMTRADA-guide för vårdpersonal Utbildningsmodul om LEMTRADA
• Checklista för förskrivare av LEMTRADA
Läs igenom produktresumén som finns tillgänglig på Europeiska läkemedelsmyndighetens webbplats enligt ovan.
Information om förberedelse för administering av LEMTRADA samt patientövervakning
• Patienterna ska premedicineras med kortikosteroider omedelbart före infusion med LEMTRADA under de första 3 dagarna av behandlingsomgången, oavsett vilken omgång det gäller. Premedicinering med antihistaminer och/eller antipyretika före administrering av LEMTRADA kan också övervägas.
• Oral behandling mot herpes ska ges till alla patienter under behandlingen och i en månad efteråt. I kliniska prövningar fick patienterna aciclovir 200 mg två gånger dagligen eller motsvarande.
• Genomför inledande tester och screening enligt beskrivning i avsnitt 4 i produktresumén.
• Innehållet i injektionsflaskan ska inspekteras före administrering med tanke på partiklar och missfärgningar. Koncentrat som är missfärgat eller innehåller partiklar får inte användas. INJEKTIONSFLASKAN FÅR INTE SKAKAS FÖRE ANVÄNDNING.
• Använd aseptisk teknik och dra upp 1,2 ml LEMTRADA från injektionsflaskan och injicera i 100 ml natriumklorid 9 mg/ml (0,9 %) infusionsvätska, lösning eller glukos 50 mg/ml (5 %) infusionsvätska, lösning. Vänd påsen försiktigt för att blanda lösningen. Var noga med att hålla den beredda lösningen steril eftersom den inte innehåller några konserveringsmedel.
• Administrera LEMTRADA infusionsvätska, lösning via intravenös administrering över cirka fyra timmar.
• Inga andra läkemedel får tillsättas till LEMTRADA infusionsvätska, lösning eller ges samtidigt genom infusion via samma intravenösa kanal.
• Det är rekommenderat att läkemedlet används omedelbart efter spädning, på grund av möjlig risk för mikrobiell kontaminering. Om läkemedlet inte används omedelbart är det användarens ansvar att se till att förvaringstiden under användning samt förhållandena före användning inte överstiger 8 timmar vid 2 °C till 8 °C, skyddat från ljus.
• Följ lämpliga rutiner för hantering och kassering. Spill och avfallsmaterial ska kasseras i enlighet med lokala krav.
• Efter varje infusion ska patienten observeras i två timmar med avseende på infusionsrelaterade reaktioner. Symtomatisk behandling kan sättas in vid behov - se produktresumén. Fortsätt kontrollera patienten för autoimmuna tillstånd varje månad tills det har gått fyra år sedan den sista infusionen. Mer information finns i LEMTRADA-guiden för vårdpersonal eller i produktresumén som finns tillgänglig på Europeiska läkemedelsmyndighetens webbplats enligt ovan.
BILAGA IV
VETENSKAPLIGA SLUTSATSER OCH SKÄL TILL ÄNDRING AV VILLKOREN FÖR GODKÄNNANDENA FÖR FÖRSÄLJNING
Vetenskapliga slutsatser
Med hänsyn till PRAC:s utredningsprotokoll om den periodiska säkerhetsrapporten för alemtuzumab är CHMP:s slutsatser följande:
Listerios/L isteria-men ingit
Användning av läkemedel med immunomodulerande effekt såsom Lemtrada kan vara förknippat med en ökad risk för opportunistiska infektioner. Sammanlagt 5 fallrapporter har identifierats, samtliga härrör från EU. En alemtuzumab-behandlad MS-patient, inkluderad i den kliniska studien CAMMS223, utvecklade listeria-meningit, och fyra spontanrapporter efter marknadsföring rör fall av antingen systemisk listerios eller Listeria monocytogenes meningit.
Bradykardi som en infusionsrelaterad biverkan
71 fall (hos 55 patienter) av bradykardi (varav två bedömdes som allvarliga, övriga icke allvarliga) rapporterades i kliniska prövningar. Totalt behandlades 1505 patienter med alemtuzumab i dessa studier. Ytterligare 39 fall av bradykardi (varav 8 bedömdes som allvarliga, övriga icke allvarliga) rapporterades efter marknadsförande från 1 maj 2015. Alla de tio allvarliga fallen med bradykardi uppträdde som infusionsrelaterade reaktioner.
I beaktande av de data som presenterats i den utvärderade periodiska säkerhetsrapporten anser PRAC att ändringar av produktinformationen för läkemedel med alemtuzumab är berättigade. Avsnitt 4.4 i produktresumén samt relevanta delar av bipacksedeln har uppdaterats.
CHMP instämmer i PRAC:s vetenskapliga slutsatser.
Skäl att ändra villkoren för godkännandet (godkännandena) för försäljning
Baserat på de vetenskapliga slutsatserna för alemtuzumab anser CHMP att nytta-riskförhållandet för läkemedlet som innehåller alemtuzumab är oförändrat under förutsättning att de föreslagna ändringarna görs i produktinformationen.
CHMP rekommenderar att villkoren för godkännandet (godkännandena) för försäljning ändras.
42